Diagnostisering og utredning av ALS gjøres av nevrolog. Diagnosen ALS er en klinisk diagnose, noe som vil si at diagnosen stilles på bakgrunn av spesifikke funn ved nevrologisk undersøkelse. Ofte vil utgangspunktet for vurderingen være kraftsvikt i et område av kroppen, og undersøkelsen vil ha et særlig fokus på å identifisere tegn til påvirkning av øvre og nedre motornevron.
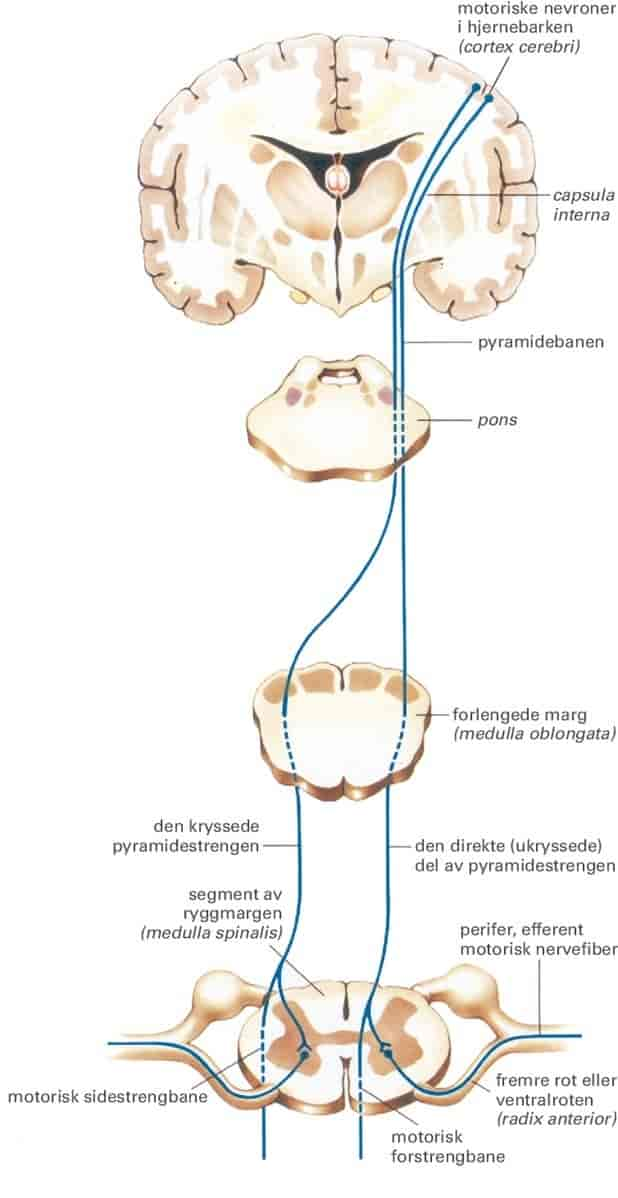
Tegn på at øvre motornevron er påvirket kan være delvis eller ufullstendig lammelse med stor temporeduksjon, livlige reflekser, unormale reflekser (som oppadvendt plantarrefleks) og økt spenning i muskulaturen (spastisitet).
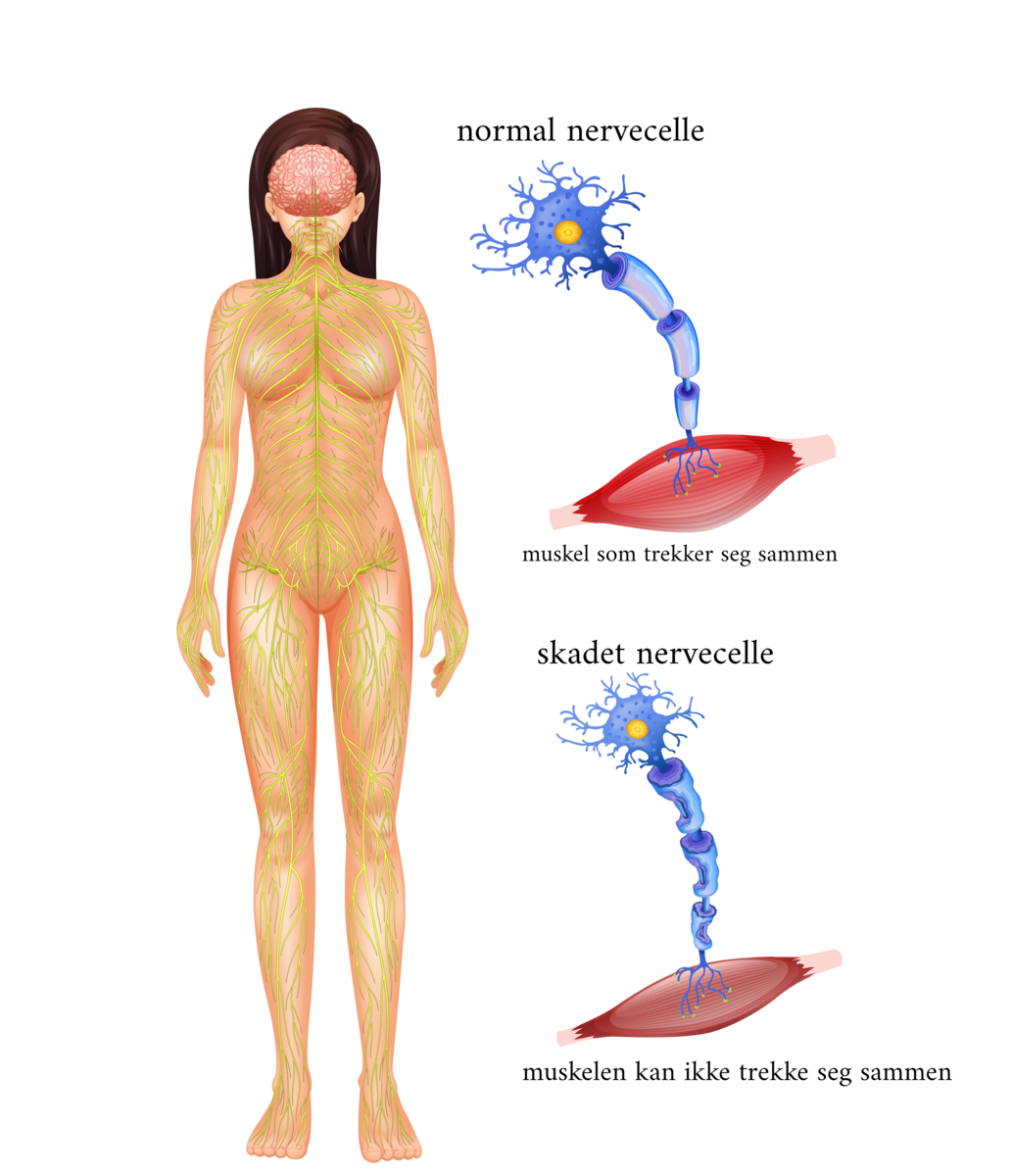
Tegn på at nedre motornevron er påvirket kan være delvis eller ufullstendig lammelse med svekkede reflekser, muskelsvinn og spesielle ufrivillige bevegelser som sees som muskelspill under huden, kalt fascikulasjoner. Nevrologen vil ofte også undersøke nedre motornevron med nevrofysiologiske undersøkelse av perifere nerver (nevrografi) og muskler (elektromyografi, EMG).
Undersøkelsene kan gi objektive mål på påvirkningen av nedre motornevron. Samtidig kan man utelukke sykdom i sensoriske nevroner som bringer sanseinformasjon fra blant annet hud og ledd til ryggmargen. Tilsvarende vil man ved nevrologisk undersøkelse avklare at andre deler av nervesystemets funksjon, særlig lillehjernen, basalgangliene og sanseapparatene ikke er rammet. Hvis man finner tegn til sykdom utenom det motoriske system, vil det som regel være nødvendig å vurdere andre nevrologiske sykdommer som årsak til symptomene.
ALS er en eksklusjonsdiagnose. Selv om legen ved den nevrologiske undersøkelsen ikke finner tegn til sykdom utenom det motoriske system, må andre årsaker til sykdomsbildet utelukkes. Da sykdommen kan starte mange steder, vil den arte seg på svært ulik vis i startfasen. Det gjør at utredningen må tilpasses den enkelte pasient. Utredning vil som regel inkludere bildeundersøkelser med MR, analyse av spinalvæske og de nevnte nevrofysiologiske undersøkelsene.
Utredningen og de diagnostiske kriteriene tar utgangspunkt i å avgrense funksjonstap til det motoriske systemet. Likevel vil en andel pasienter utvikle kognitive endringer som skyldes sykdom i større områder av pannelappen enn i den motoriske hjernebarken. Dette er ikke samme type endringer som ved andre nevrodegenerative sykdommer, som for eksempel demens av Alzheimer type. Pasienter med ALS kan få personlighetsendring hvor apati, initiativløshet og beslutningsvansker er vanlig. Hos omtrent 15 prosent av pasientene med ALS utvikler det seg en full pannelappdemens, mens en større andel pasienter kan ha lette endringer. Det er likevel mange pasienter med ALS som ikke opplever endringer i kognitiv funksjon.
Sykdommen kan i utgangspunktet ramme kontroll av all viljestyrt muskulatur. Likevel er det et gjennomgående trekk at tverrstripet muskulatur som styrer øyebevegelser og endetarmens lukkemuskel rammes svært sent i forløpet, og i de fleste sykdomsforløp vil ikke funksjonstap vise seg her. Mange pasienter vil derfor kunne styre kommunikasjonshjelpemidler med viljestyrte øyebevegelser, også sent i sykdomsforløpet.



Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.