
Fenylketonuri er en medfødt, arvelig stoffskiftesykdom som skyldes mangel på enzymet fenylalanin-hydroksylase.
Faktaboks
- Uttale
- fenˈylketonurˈi
- Også kjent som
-
oligophrenia phenylpyruvica, Føllings sykdom, phenylketonuri, PKU
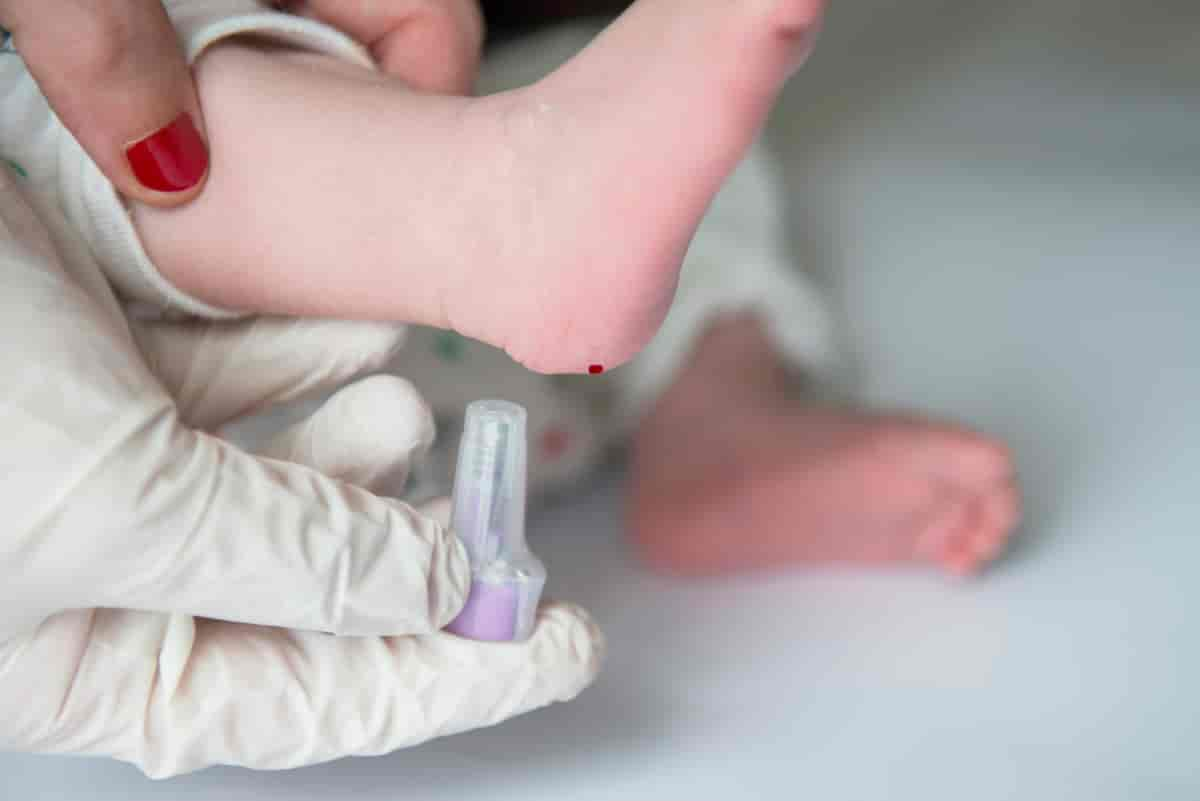
I Norge tas det en blodprøve av alle nyfødte for å undersøke om barnet har stoffskiftesykdommer.
Fenylketonuri er en medfødt, arvelig stoffskiftesykdom som skyldes mangel på enzymet fenylalanin-hydroksylase.
oligophrenia phenylpyruvica, Føllings sykdom, phenylketonuri, PKU
Enzymet fenylalanin-hydroksylase omdanner aminosyren fenylalanin til tyrosin. Fenylalanin hoper seg opp i kroppen. Omdannelsesprodukter av fenylalanin skilles ut i urinen som fenylpyrodruesyre.
Det høye nivået av fenylalanin fører til redusert transport av andre aminosyrer som er nødvendige for normal utvikling av hjernen over blod-hjernebarrieren. Resultatet er hjerneskade og gradvis økende funksjonshemning.
I Norge undersøkes alle nyfødte for fenylketonuri gjennom nyfødtscreeningen.
Friske arvebærere kan ofte identifiseres ved en fenylalaninbelastningsprøve eller ved genetisk testing, noe som åpner en viss mulighet for forebygging.
Behandlingen består i å la barnet så tidlig som mulig få mat med lite fenylalanin.
Hvis fenylketonuri ikke behandles, får barnet en alvorlig hjerneskade og sterk grad av psykisk utviklingshemning. Ved riktig behandling kan barnet få en normal eller nesten normal intelligensutvikling. Andre symptomer kan også forsvinne.
Sykdommen blir også kalt Føllings sykdom etter den norske legen og professoren Asbjørn Følling (1888–1973).

Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.