
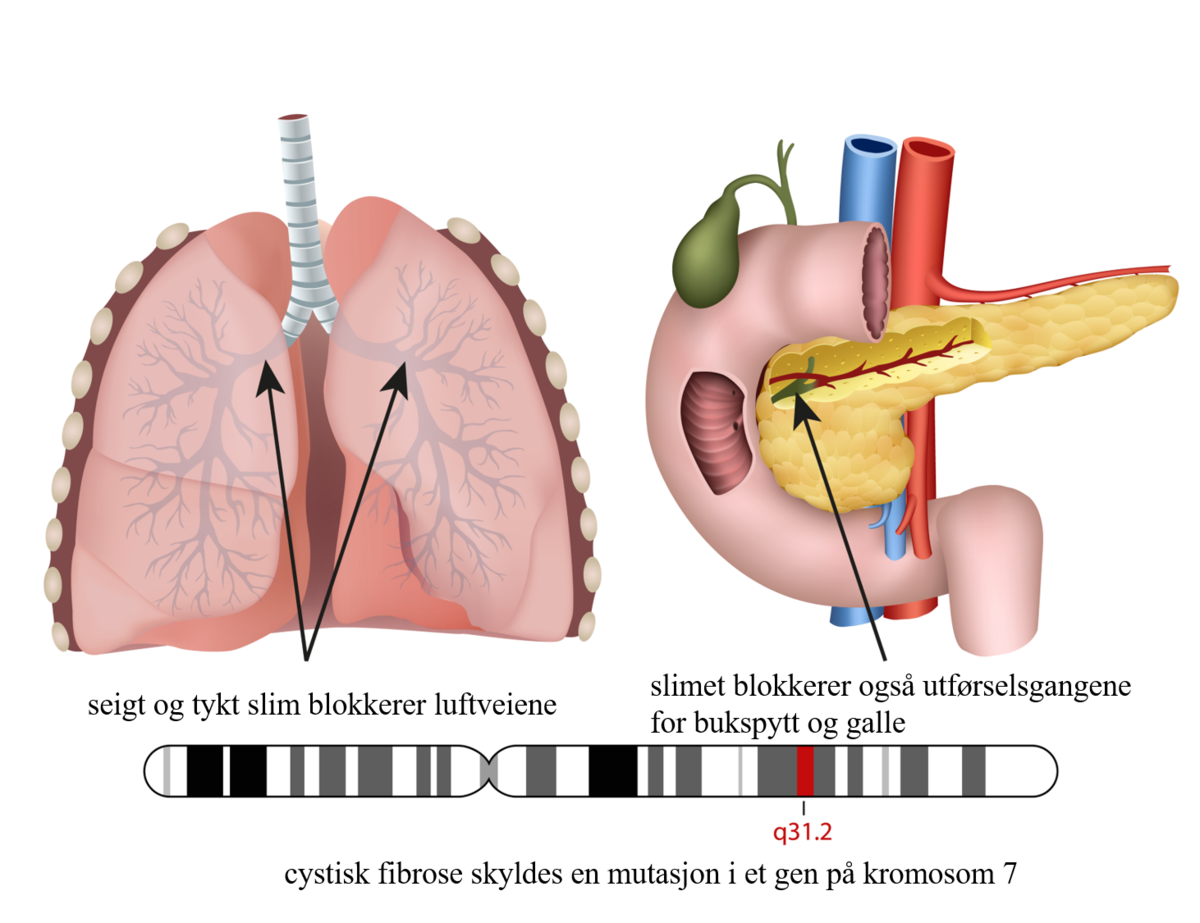
Cystisk fibrose er en medfødt sykdom som gir symptomer fra lungene og mage-tarmkanalen. Tilstanden skyldes en genfeil. Det finnes mange forskjellige genfeil som gir denne sykdommen. Anslagsvis 1 av 4000 barn har cystisk fibrose, mens 1 av 30 er friske bærere av arveanlegget.
Faktaboks
- Uttale
- sˈystisk fibrˈose
Sykdommen kjennetegnes av hoste med gulgrønt slim og stadige luftveisinfeksjoner. Det er også vanlig at pasientene sliter med å ta opp næringen fra tarmen (malabsorpsjon).
Lungefysioterapi er en viktig del av behandlingen. Det finnes en egen forening for pasienter og pårørende kalt Norsk forening for cystisk fibrose.

Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.