



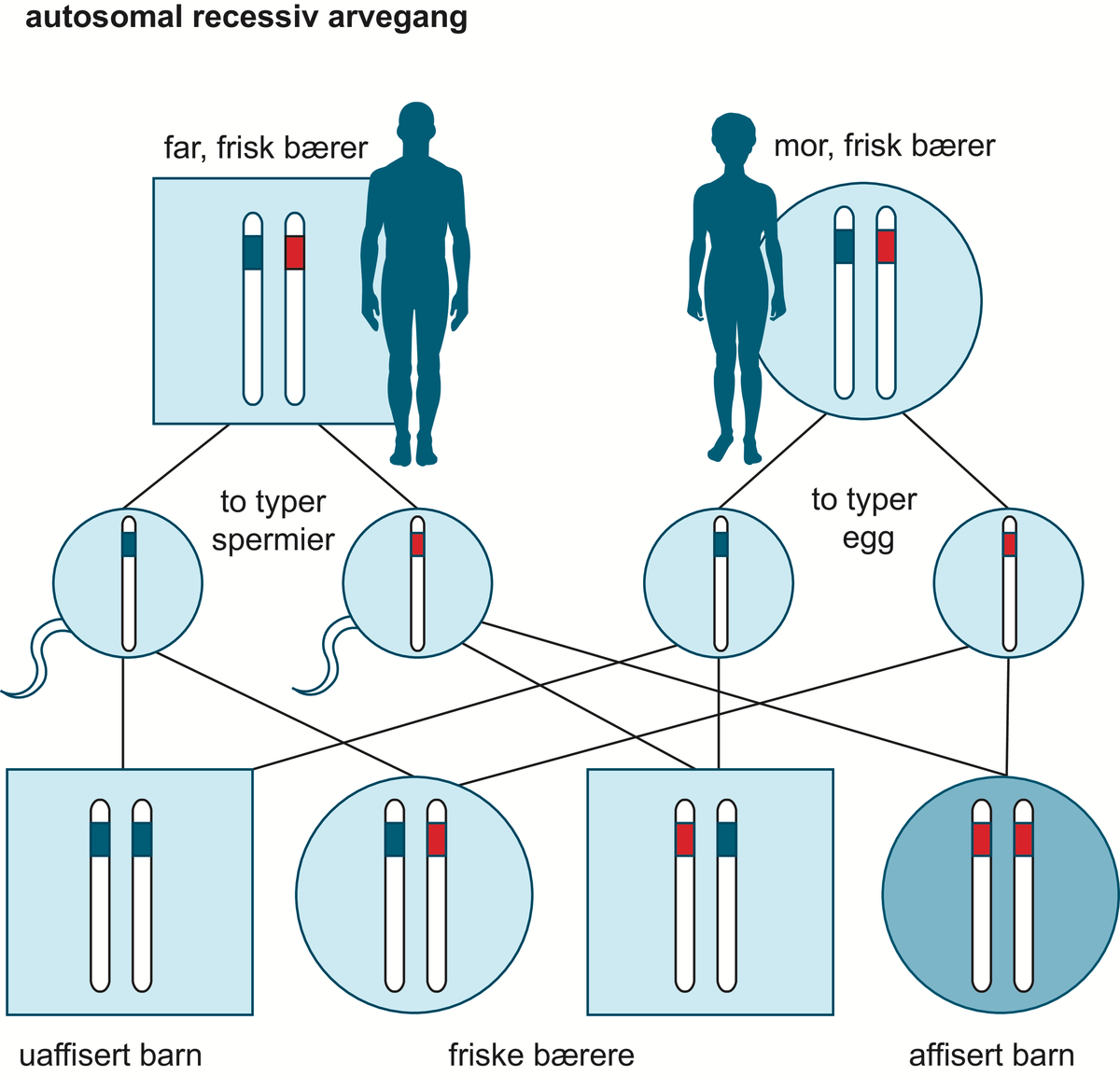
Albinisme er ein arveleg tilstand med medfødde utviklingsforstyrringar i auga og oftast mangel på fargestoffet melanin i hud, hår og auge. Pigmentmangelen fører til at ein person med albinisme er meir sårbar for ultrafiolett stråling frå sola, og gir høg risiko for hudkreft og andre hudforandringar.
Det finst to hovudformer:
- okulokutan albinisme (av latin oculus, 'auge' og cutis, 'hud'), som rammar hud, hår og auge
- okulær albinisme, som berre gir symptom frå auga
Okulokutan albinisme er mest vanleg.
Albinisme kan klassifiserast både som ein arveleg, medfødd stoffskiftesjukdom og som eit medfødd utviklingsavvik i auga. Det kan òg sjåast som del av nokre svært sjeldne, genetiske syndrom som har ei anna genetisk årsak, og der albinisme berre er eitt av fleire trekk.

{kind=link}
{kind=link}
Kommentarar
Kommentarar til artikkelen blir synleg for alle. Ikkje skriv inn sensitive opplysningar, for eksempel helseopplysningar. Fagansvarleg eller redaktør svarar når dei kan. Det kan ta tid før du får svar.
Du må være logga inn for å kommentere.