
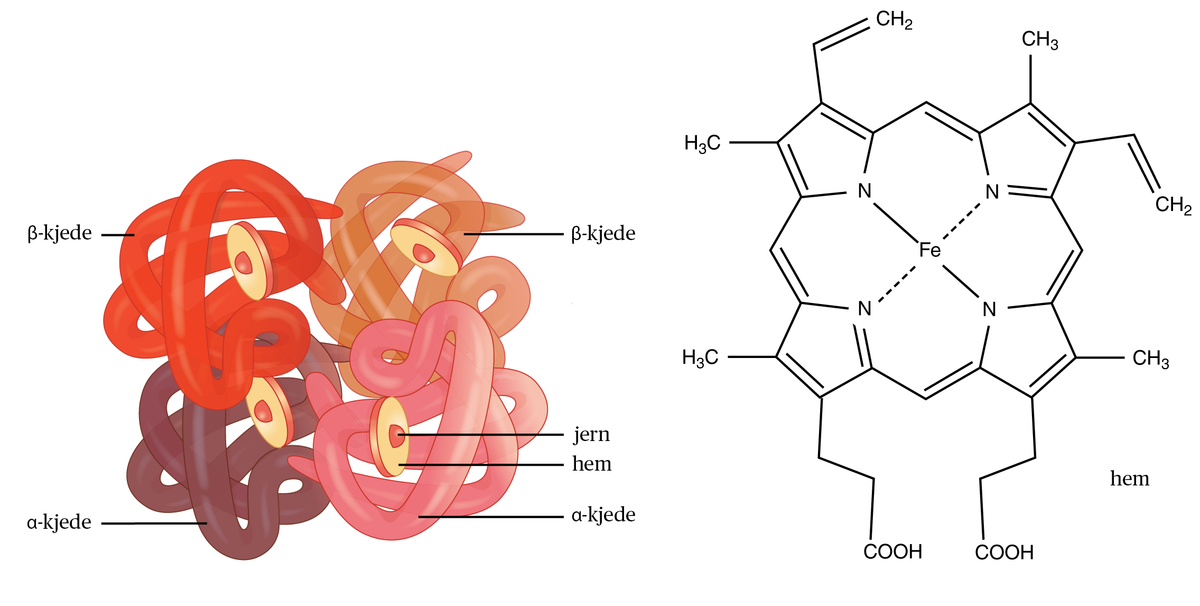
Talassemi er en arvelig blodsykdom som rammer hemoglobinet i de røde blodcellene og gir anemi. Talassemi er mest utbredt i områdene rundt Middelhavet. De vanligste symptomene er anemisymptomene: Huden blir blekere, man føler seg slappere og blir mer tung i pusten.
Sykdommene opptrer med mange alvorlighetsgrader. Den alvorligste formen kan føre til at fosteret dør i mors liv, mens den lette og vanligste formen ofte ikke merkes. Den viktigste komplikasjonen utenom anemien er avleiring av jern i organer som lever, endokrine kjertler og hjertet. Dette finnes det i dag behandling for.


Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.