
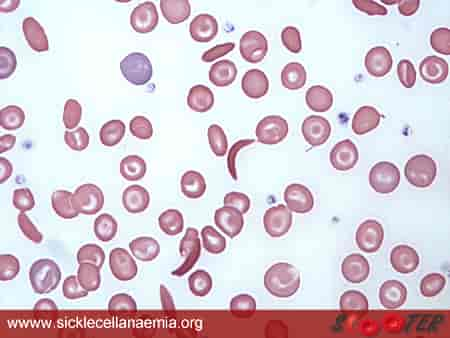

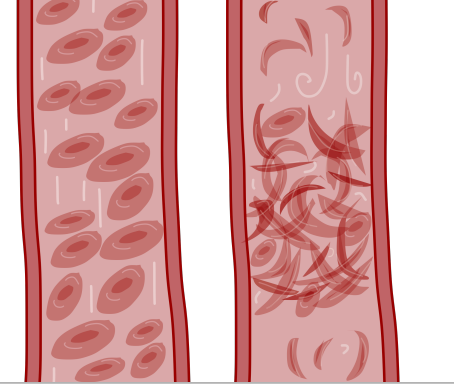
Sigdcelleanemi er en alvorlig blodsykdom som er arvelig. Det er den mest alvorlige sykdommen i en gruppe av arvelige sykdommer som omtales som sigdcellesykdom. Felles for disse er at de røde blodcellene blir sigdformede. Dette kan man bare se hvis man ser på blodcellene gjennom et mikroskop. Når blodcellene har denne formen går de lettere i stykker (hemolytisk anemi). Formen på cellene gjør også at de lettere kan forårsake blodpropper. Det gir sirkulasjonsforstyrrelser med symptomer og skader fra mange organer i kroppen.
Faktaboks
- Også kjent som
-
engelsk sickle cell anemia
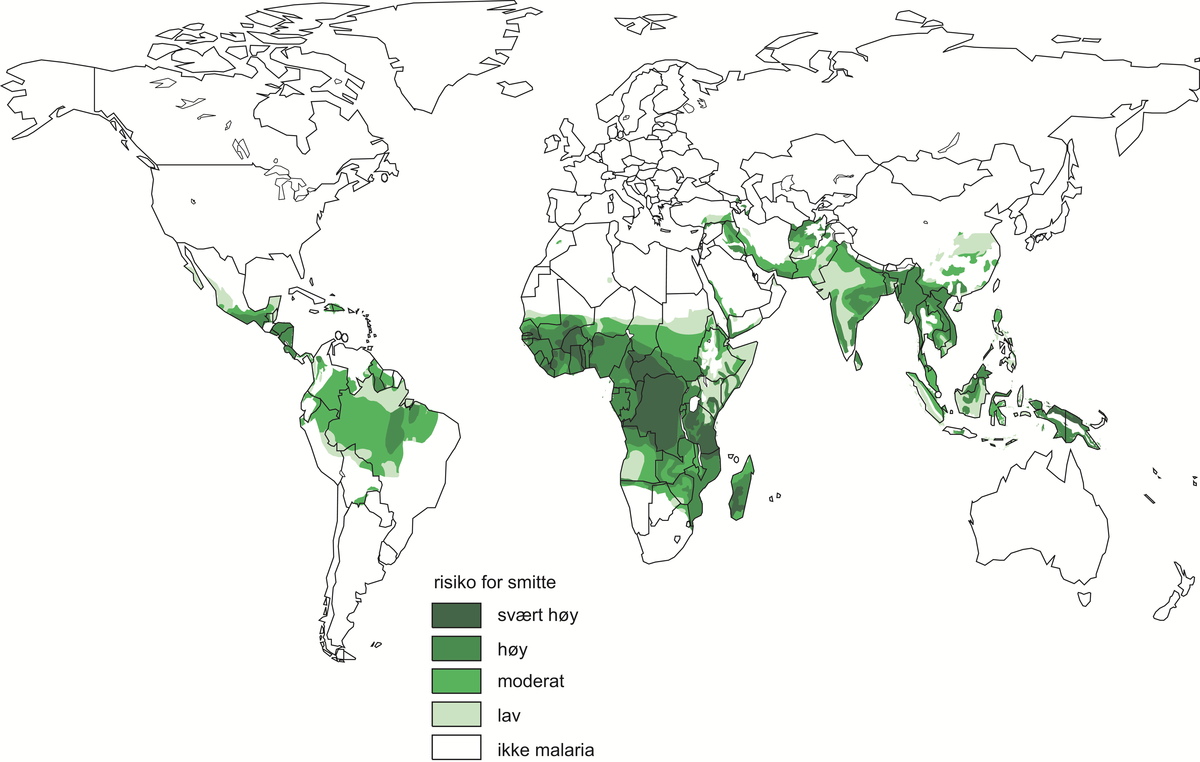
Sigdcelleanemi er blant de mest vanlige arvelige sykdommene i verden. Den forekommer særlig i sentrale deler av Afrika, Midtøsten, rundt Middelhavet og i India. Utbredelsen skyldes at sykdommen gir beskyttelse mot malaria. Evolusjonsmessig er det derfor gunstig å ha en viss grad av sigdceller i områder med malaria. Med økende folkevandring er forekomsten økt også i andre deler av verden.
Sigdcelleanemi er en alvorlig kronisk sykdom med økt dødelighet. Bærertilstanden gir lite symptomer og er ikke farlig.

Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.