
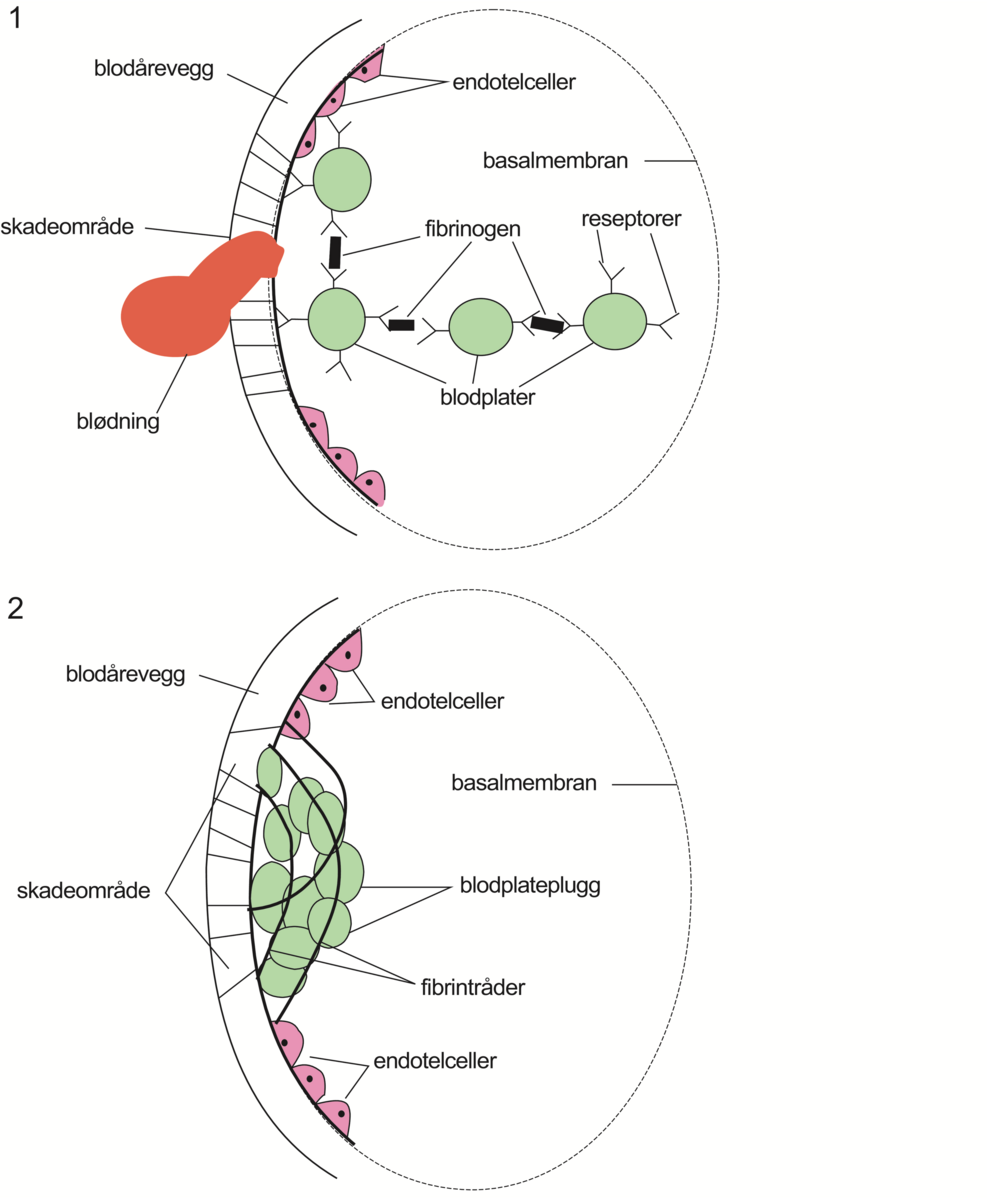
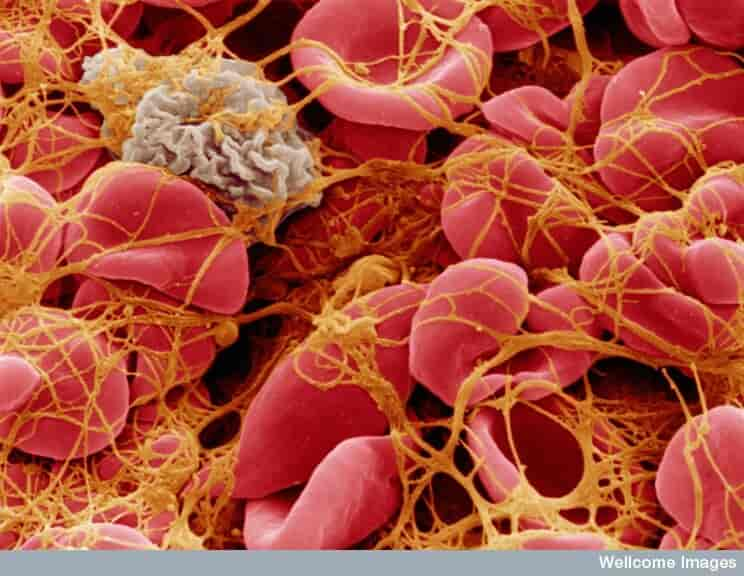
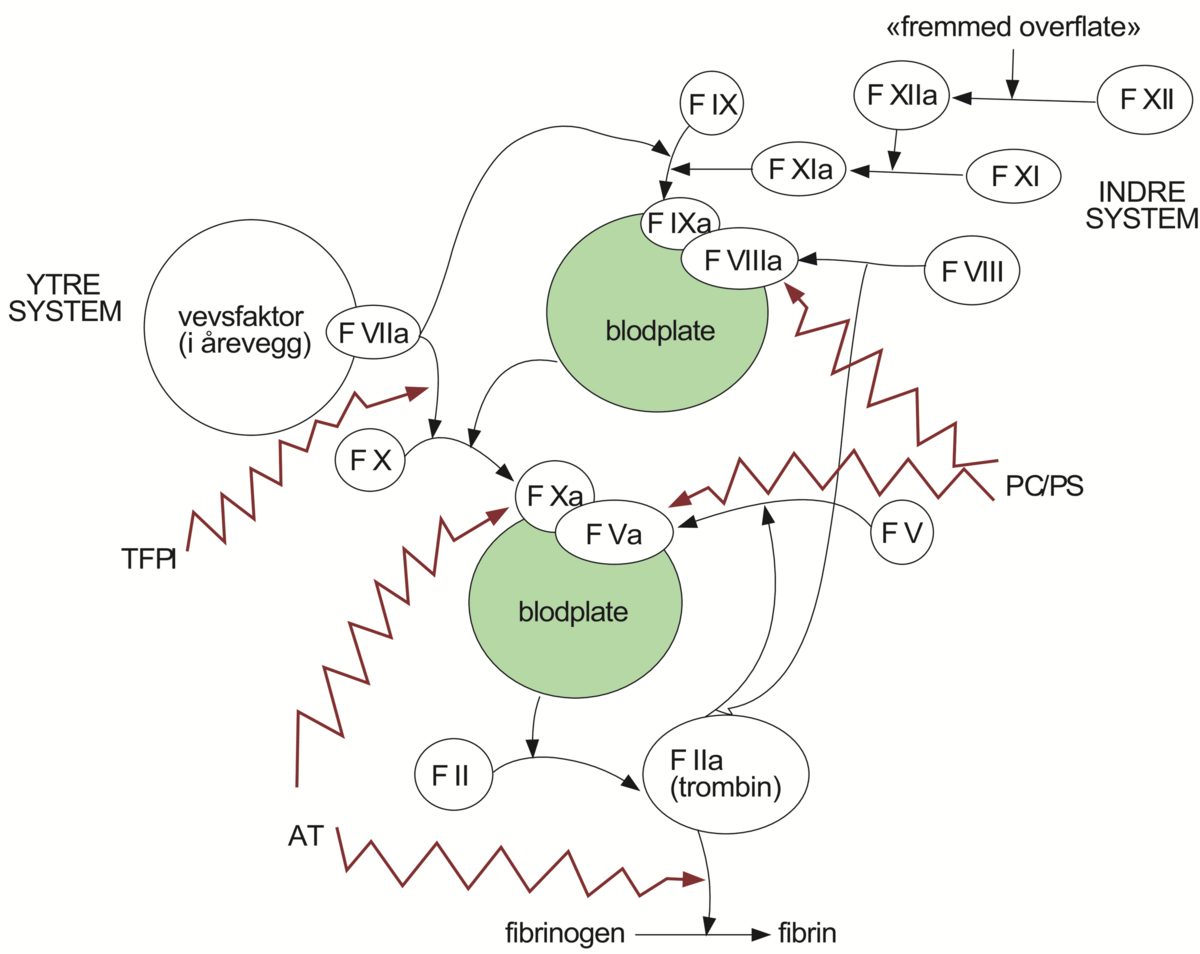
Koagulasjon er prosessen der blod klumper seg (koagulerer). Koagulasjon er blodets egen måte å stanse blødninger på.
Selve koagulasjonen er en komplisert prosess som involverer blodåreveggen, blodplater og proteiner i blodet kalt koagulasjonsfaktorer. Hvis blodåreveggen skades, vil blodplatene feste seg her og danne en såkalt blodplateplugg. Dette stanser midlertidig blødningen. Koagulasjonsfaktorene er enzymer som kan bli aktivert av blodplatene. Koagulasjonsfaktorene kan lage et nett som legger seg rundt blodplatepluggen og forsterker denne. Også røde blodceller blir «fanget» i dette nettet. Blodplatepluggen med nettet kalles et koagel.
Hvis det blir for mye koagulasjon, kan det oppstå blodpropp (trombose). Hvis det blir for lite koagulasjon, kan man blø lettere, det vil si økt blødningstendens.
Begrepet koagulasjon brukes også i videre forstand om prosesser der partikler eller molekyler slutter seg sammen. Se koagulering.
Kommentarer (1)
skrev Karoline Sætre
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.